上一篇我们把 LUAD 的预后骨架搭好了:13 基因 LASSO 风险评分,在 504 例 TCGA-LUAD 患者上能稳定区分高低风险(log-rank p << 0.001),并在多因素 Cox 中保持独立预后意义。但仅有一个评分还不够支撑一篇『故事完整』的预后论文。本篇会沿着 7 条独立的轴线展开,每一条都对应论文中的一个独立 Figure:(1)WGCNA 找与 risk 相关的共表达模块;(2)肿瘤免疫微环境与免疫检查点;(3)Hallmark 50 通路的单样本活性;(4)分子亚型(无监督共识聚类);(5)模型可信度评估(C-index / DCA / 随机生存森林);(6)miRNA-mRNA 调控网络;(7)驱动突变景观与样本流向。
第六步 · WGCNA:把上千个基因压成 10 个模块
WGCNA 的核心动作只有两步:① 选合适的 soft-threshold 让基因相关性矩阵近似 scale-free;② 通过 TOM 距离 + dynamic tree cut 把基因聚成模块,再用 module eigengene(每个模块的代表)与临床性状做相关。我们在 top-5000 MAD 基因上构网,最终聚出 10 个模块。
sft <- pickSoftThreshold(datExpr, networkType = "signed",
powerVector = c(1:10, seq(12, 20, 2)))
net <- blockwiseModules(datExpr, power = sft$powerEstimate,
networkType = "signed", TOMType = "signed",
minModuleSize = 30, mergeCutHeight = 0.25)
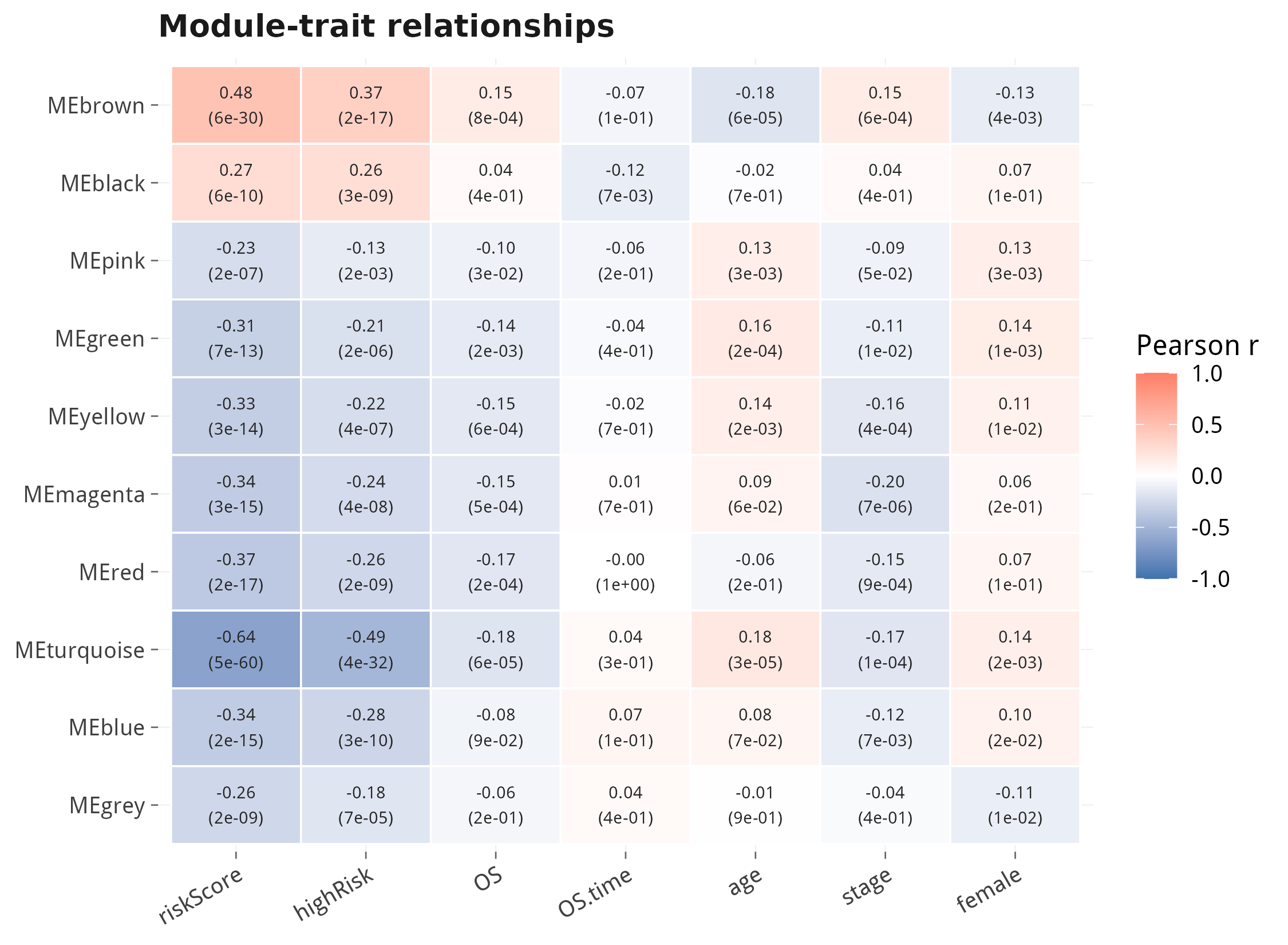
modCor <- cor(MEs, traits) # module x trait correlation6.1 模块识别:基因聚类树 + 颜色条
6.2 模块-性状相关热图(核心结果)
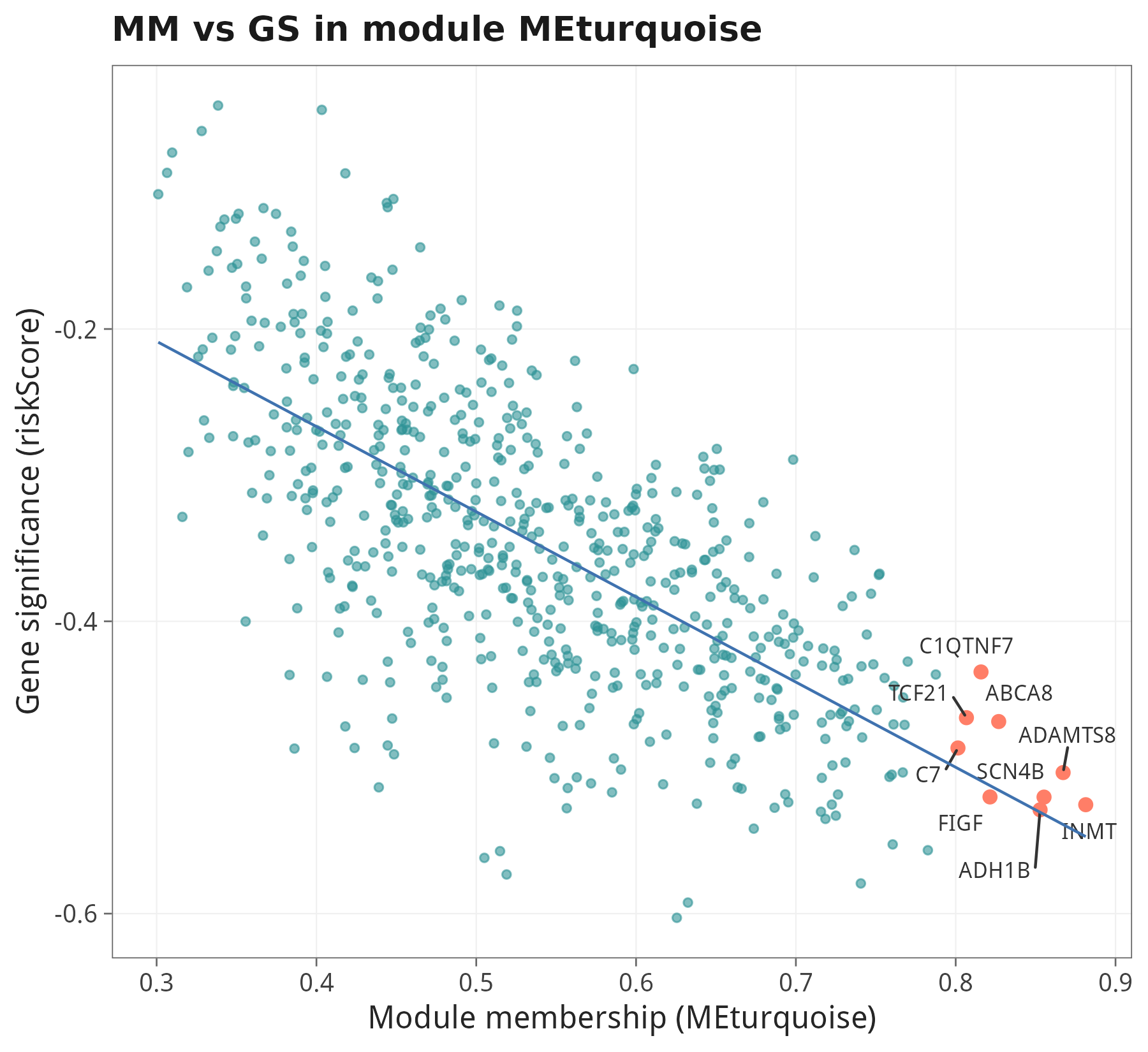
6.3 Hub gene:MM × GS 双轴筛选
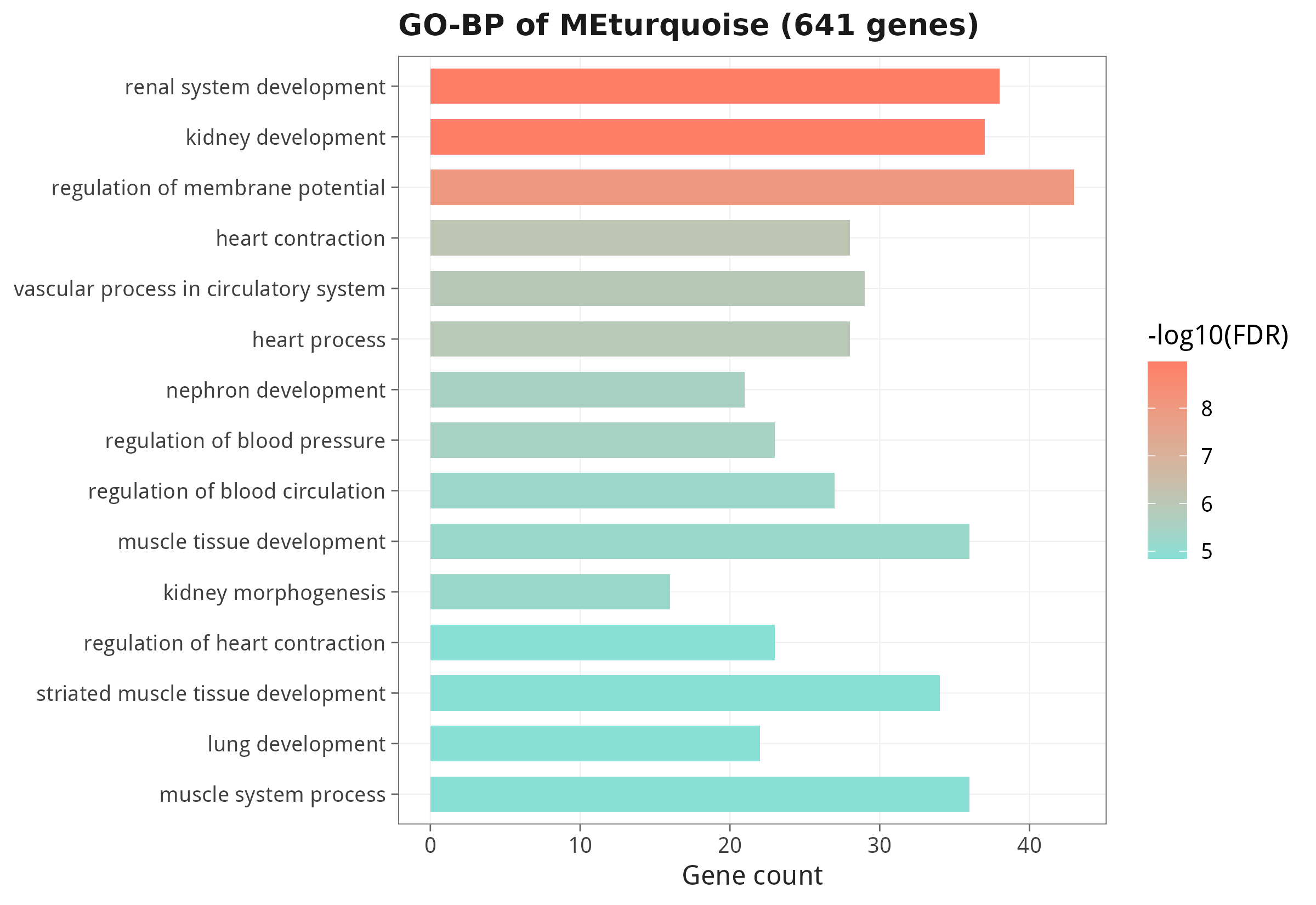
6.4 关键模块的 GO-BP 功能
第七步 · 肿瘤免疫微环境(ESTIMATE + ssGSEA + 检查点)
TIME 我们从三个角度切入:(a)ESTIMATE 用 141 个基因签名估 stromal / immune / 综合得分,顺带导出 tumor purity;(b)ssGSEA 在 Bindea 23 套免疫细胞签名上给每个样本打分;(c)22 个免疫检查点基因(PD1, PD-L1, CTLA-4, BTLA, TIGIT, …)分别与 risk score 算相关 / 与 risk group 做差异。
# ESTIMATE
filterCommonGenes(input.f = expr_in, output.f = cg, id = "GeneSymbol")
estimateScore(input.ds = cg, output.ds = scoreFile, platform = "illumina")
# ssGSEA on Bindea 23 immune cell signatures
gp <- ssgseaParam(exprData = mrna_t, geneSets = bindea, normalize = TRUE)
ssg <- gsva(gp)7.1 ESTIMATE 三种评分:High vs Low risk
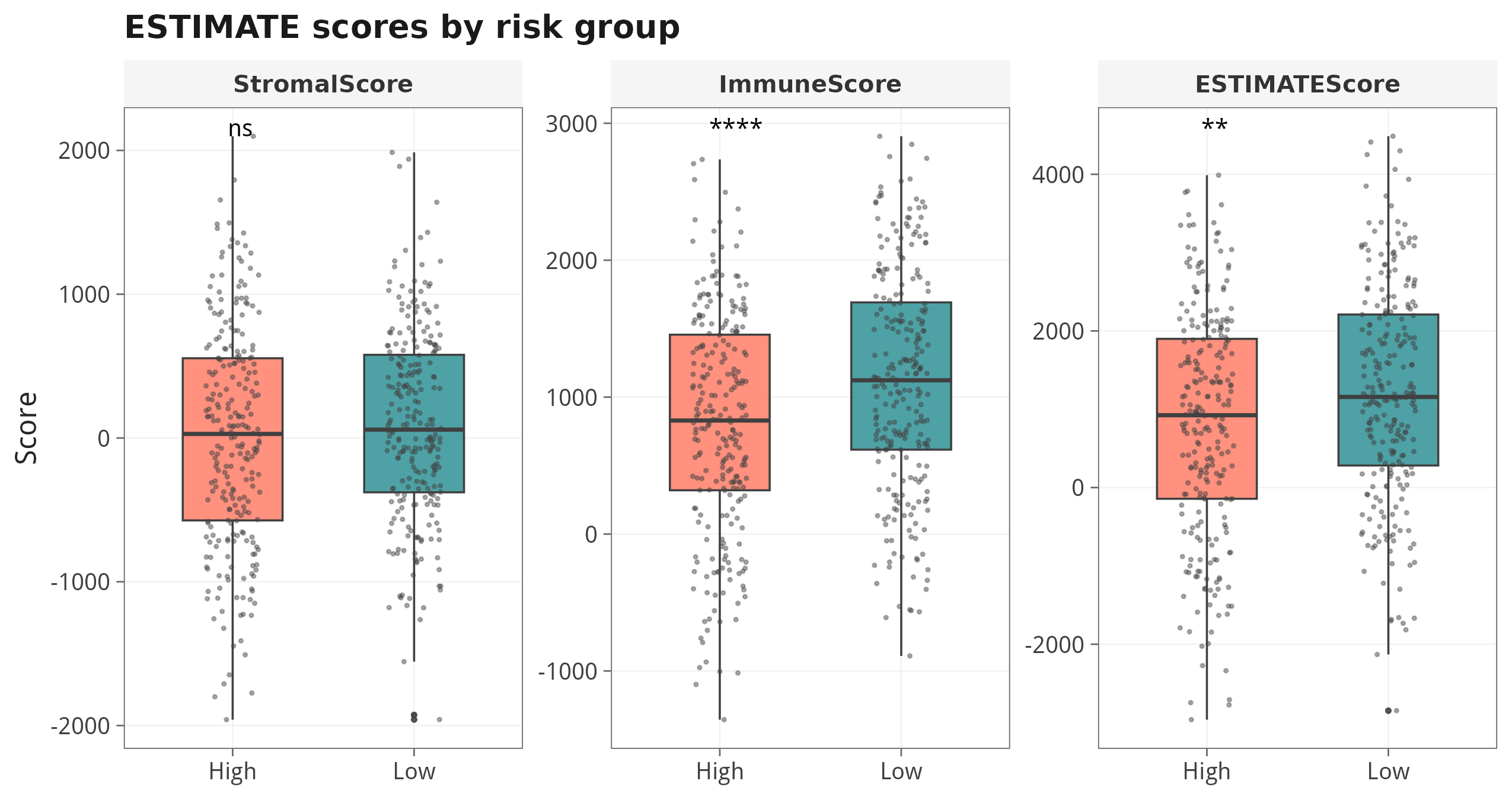
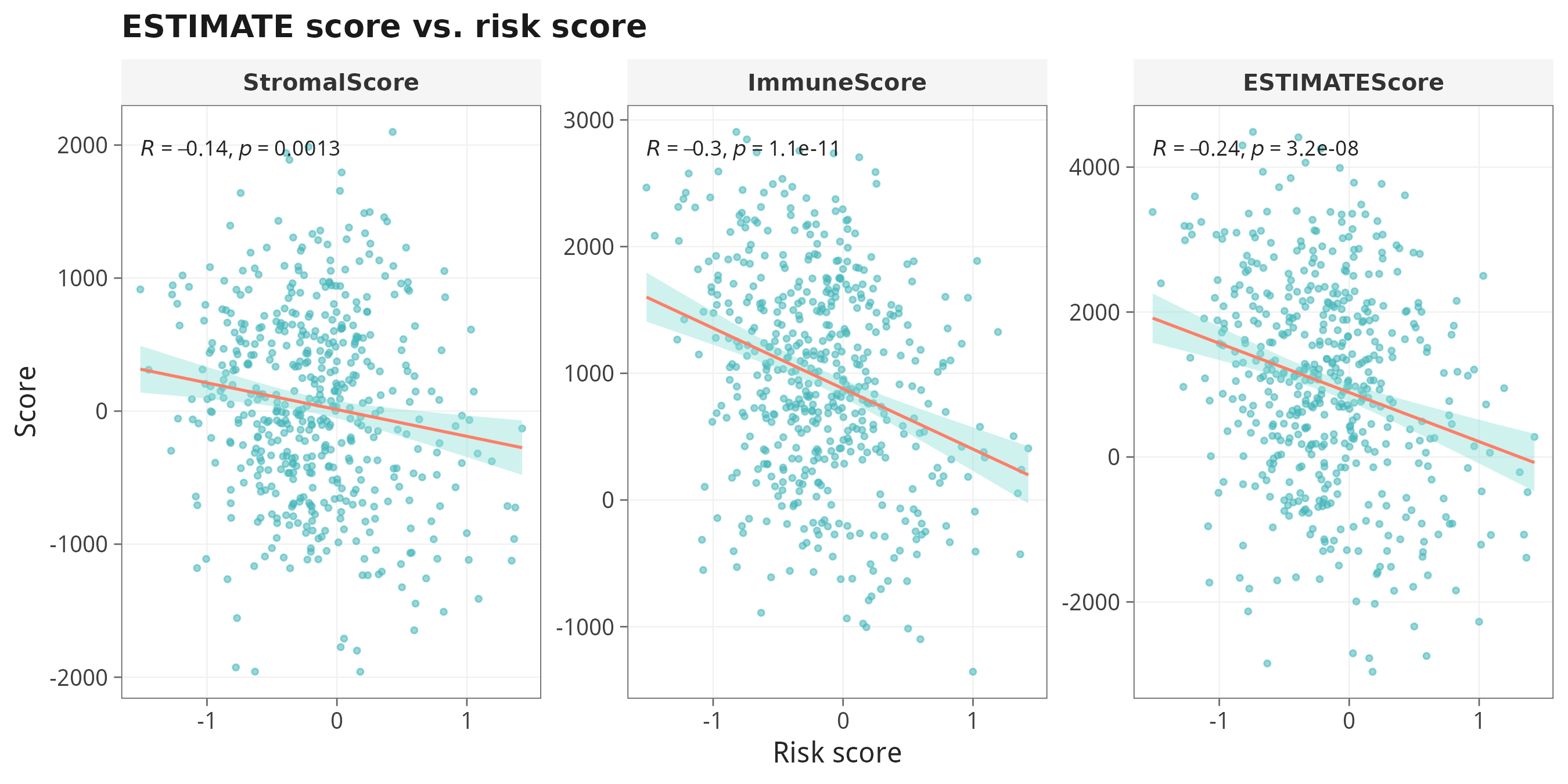
7.2 ESTIMATE vs Risk score 连续相关
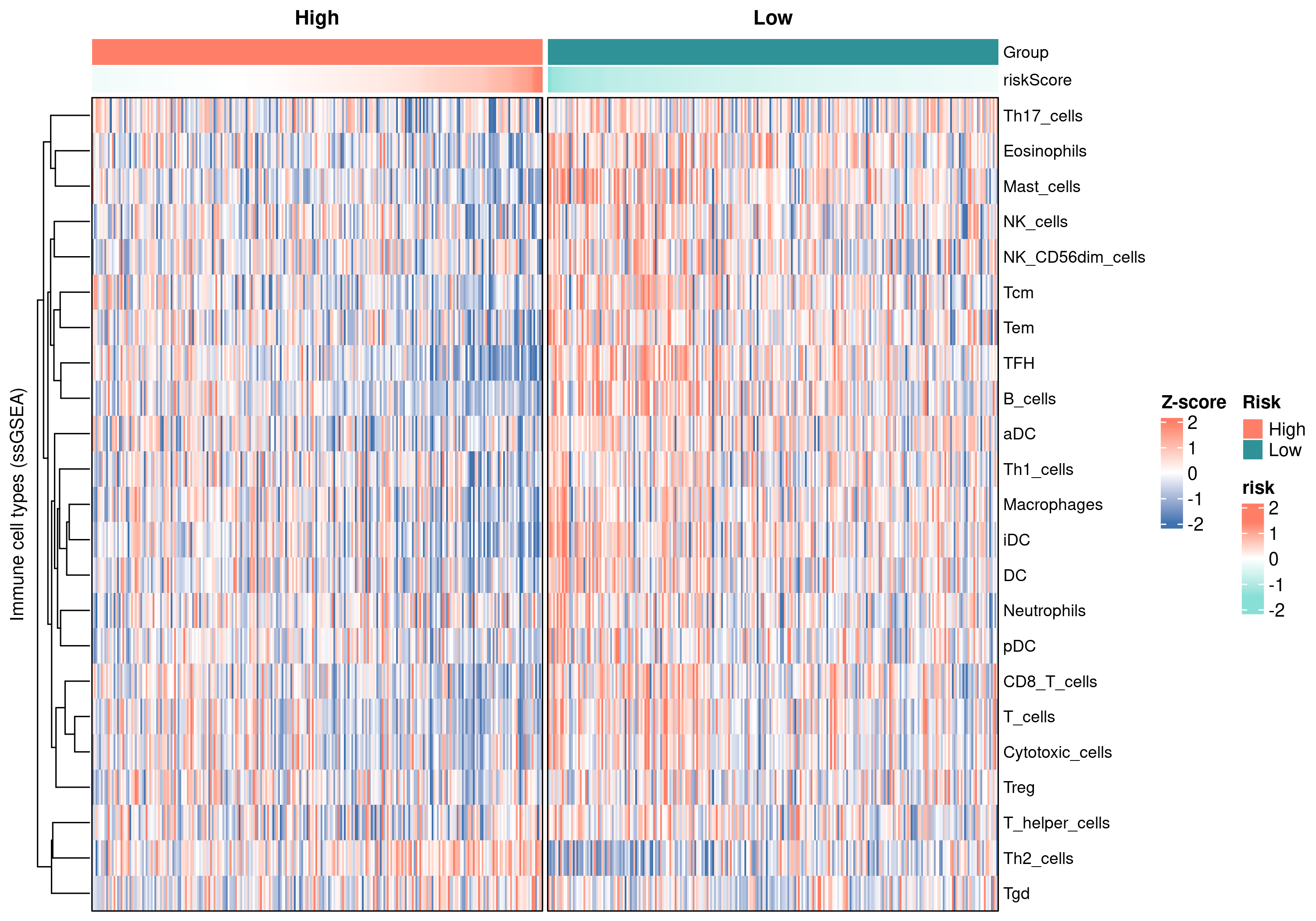
7.3 ssGSEA 23 套免疫细胞签名热图
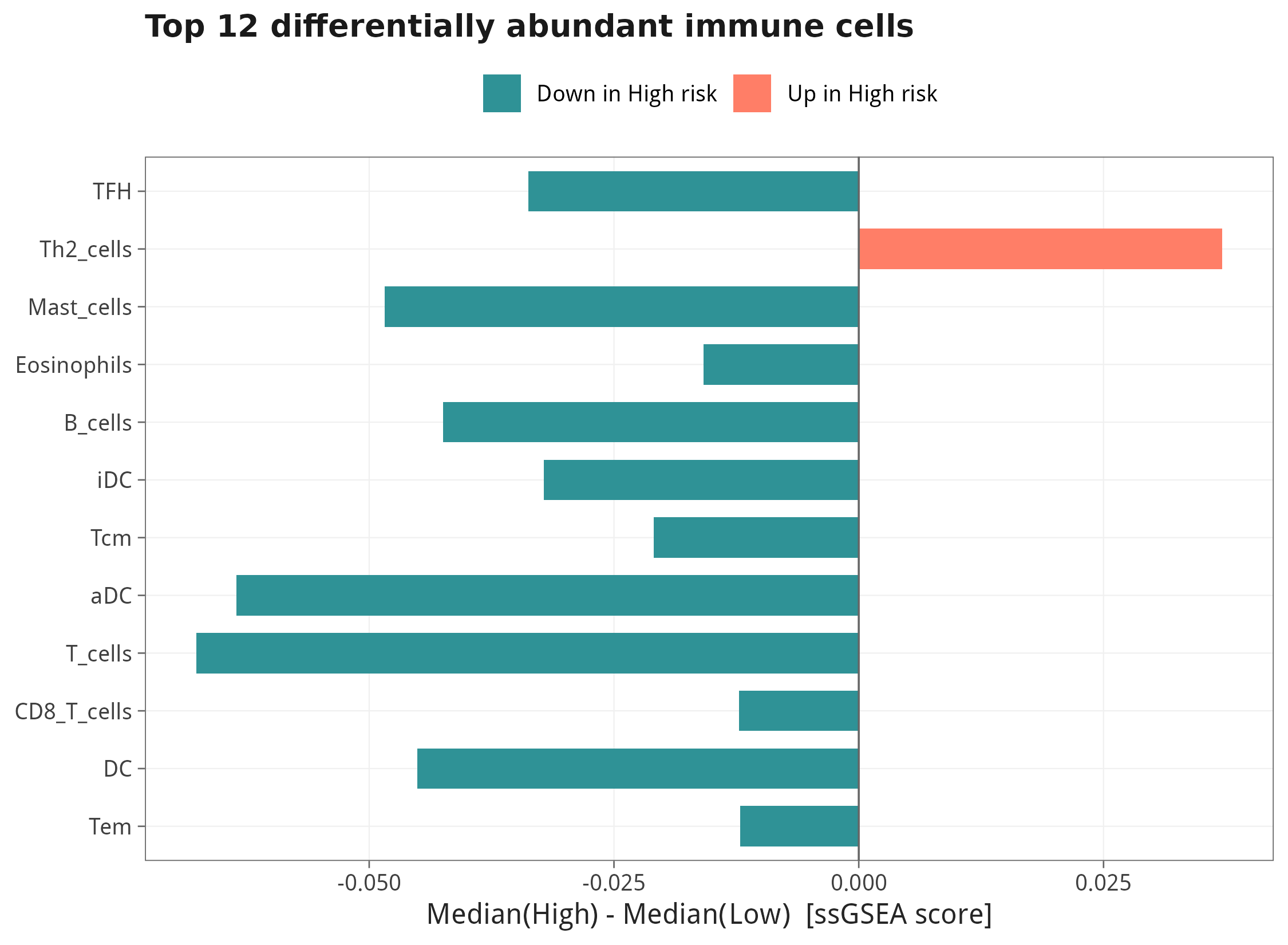
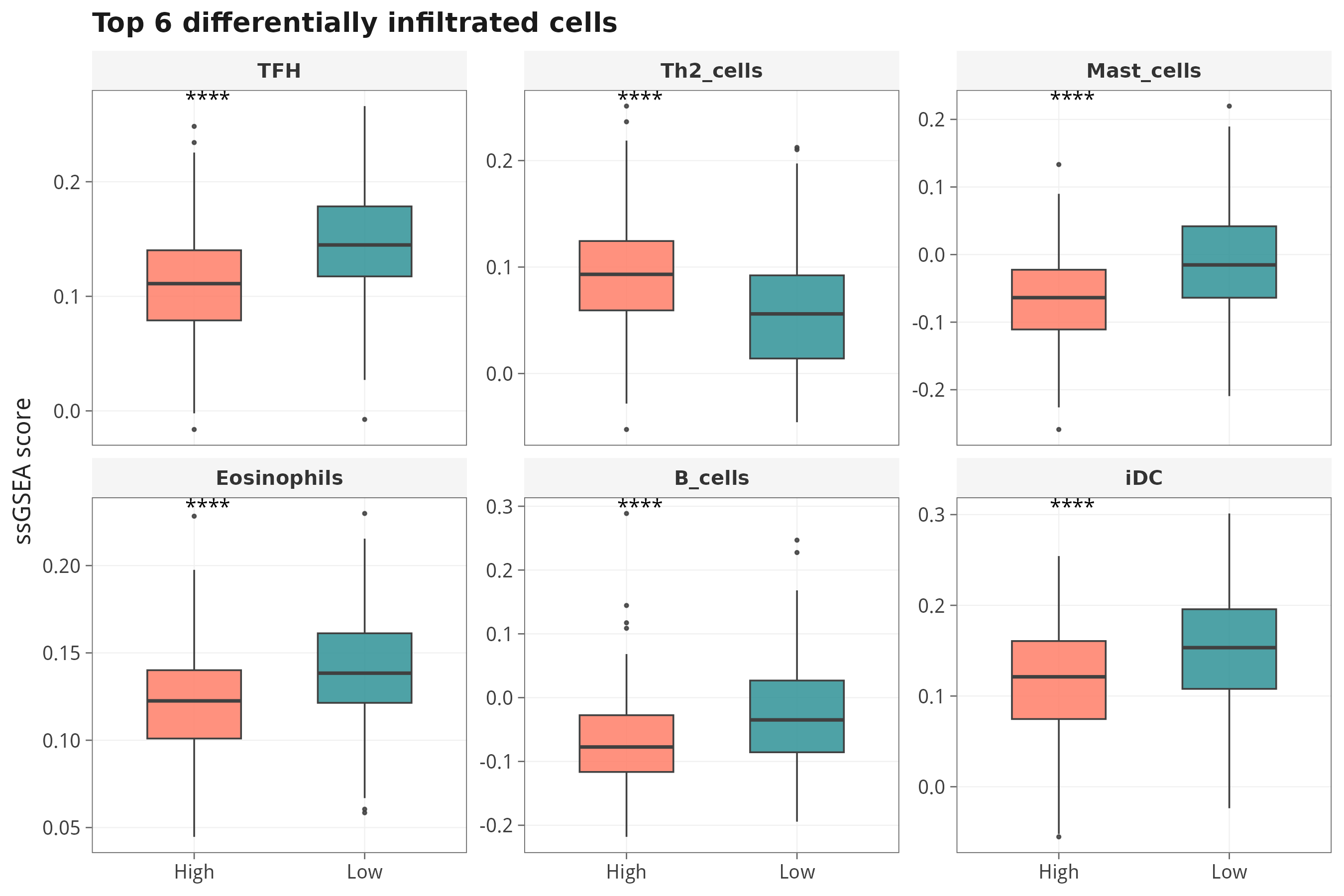
7.4 差异最显著的免疫细胞 Top 12 / Top 6
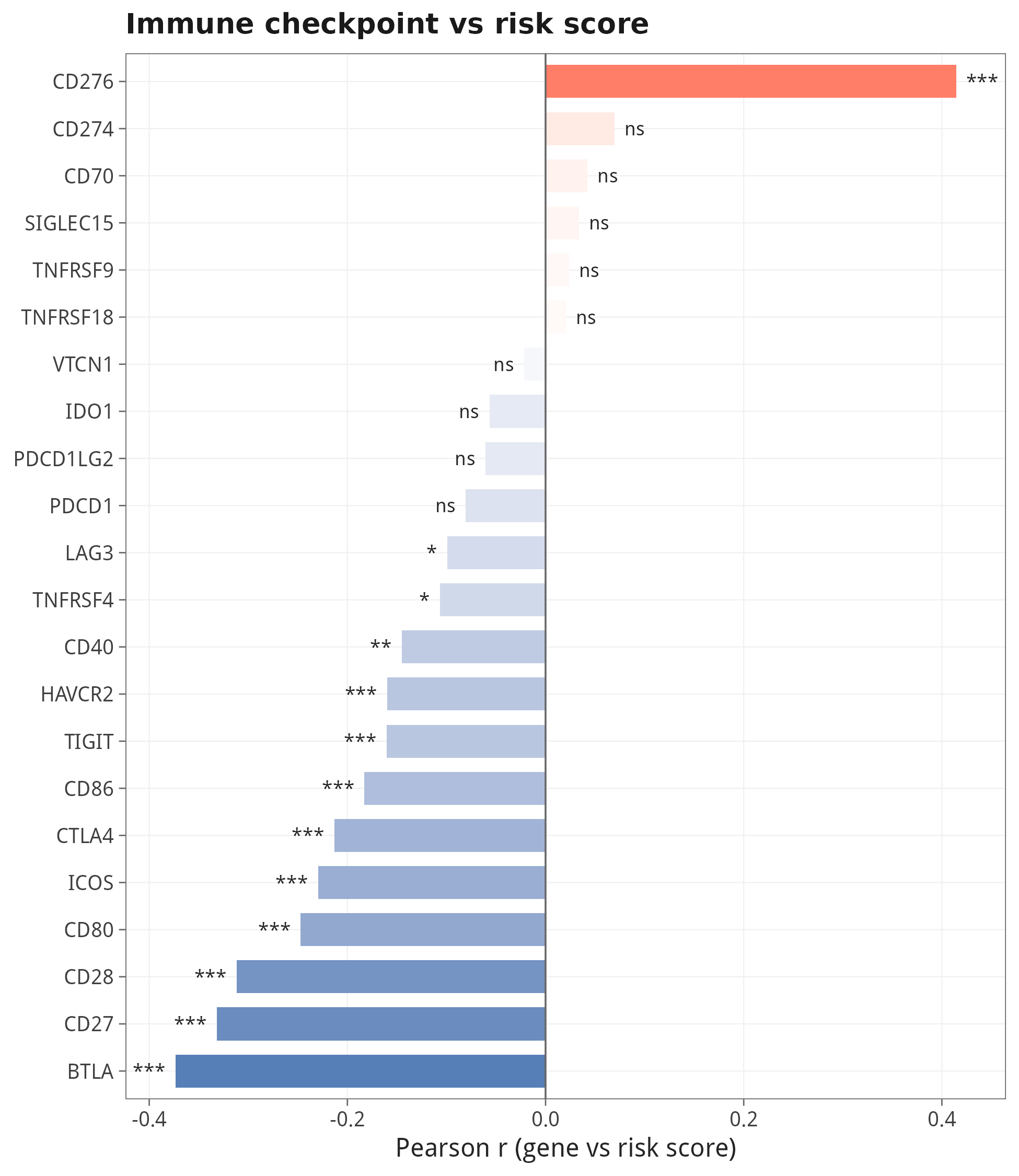
7.5 免疫检查点基因与 risk score 的相关性
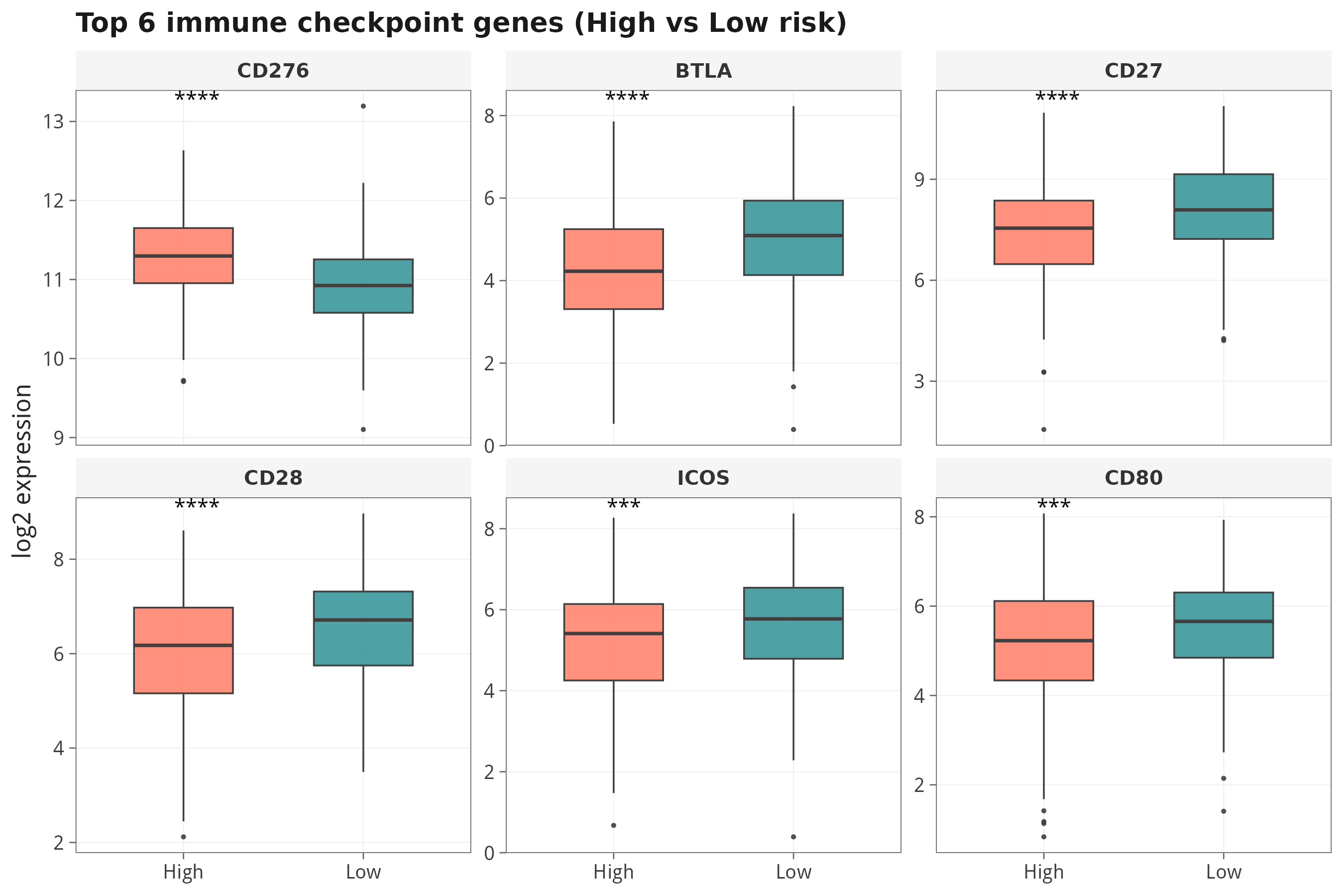
第八步 · Hallmark 50 通路的单样本活性(GSVA / ssGSEA)
ssGSEA 给每个样本一个 pathway activity vector,再用 limma 跑高低风险组的差异通路,比传统 GSEA 多两个优势:(1)每位患者都有个体水平的通路分数;(2)可以直接放进相关 / Cox / 聚类等其他分析。
hallmark <- msigdbr(species = "Homo sapiens", category = "H")
gp <- ssgseaParam(mrna_t, hallmark_geneSets, normalize = TRUE)
ssg <- gsva(gp)
design <- model.matrix(~ risk_group)
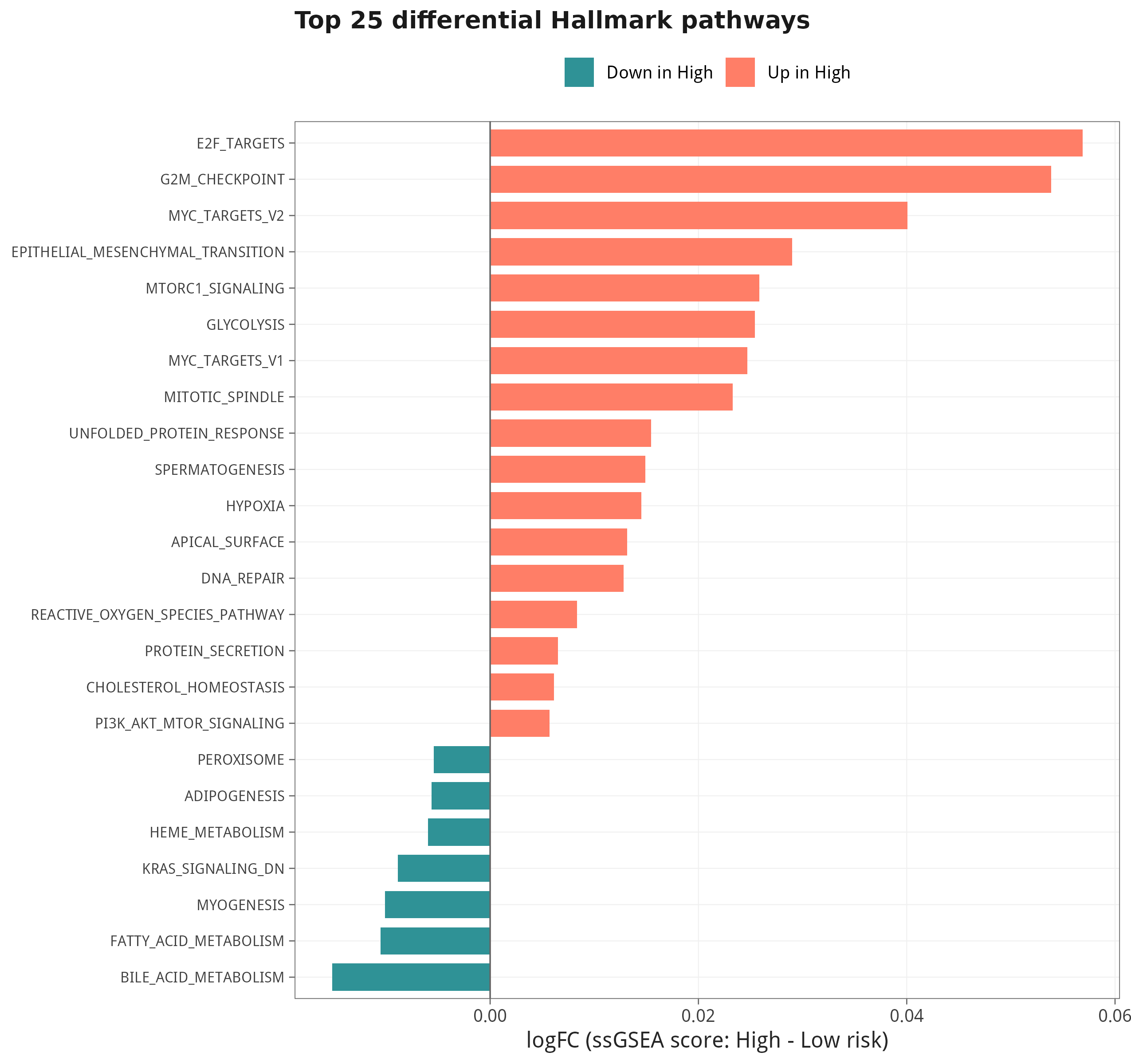
fit <- eBayes(lmFit(ssg, design))8.1 差异最显著的 Hallmark 通路
8.2 整体热图
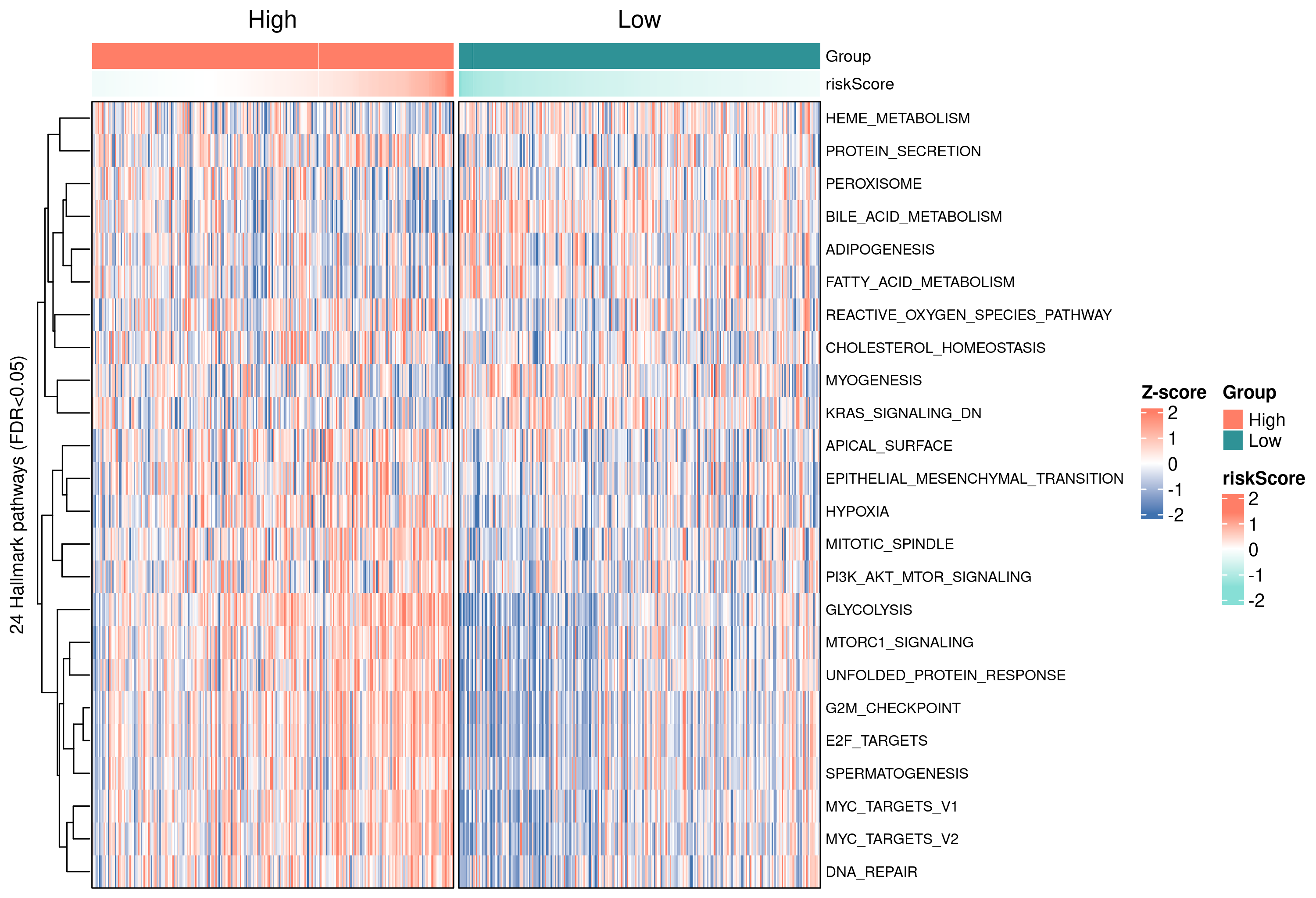
8.3 与 risk score 相关性 Top 20
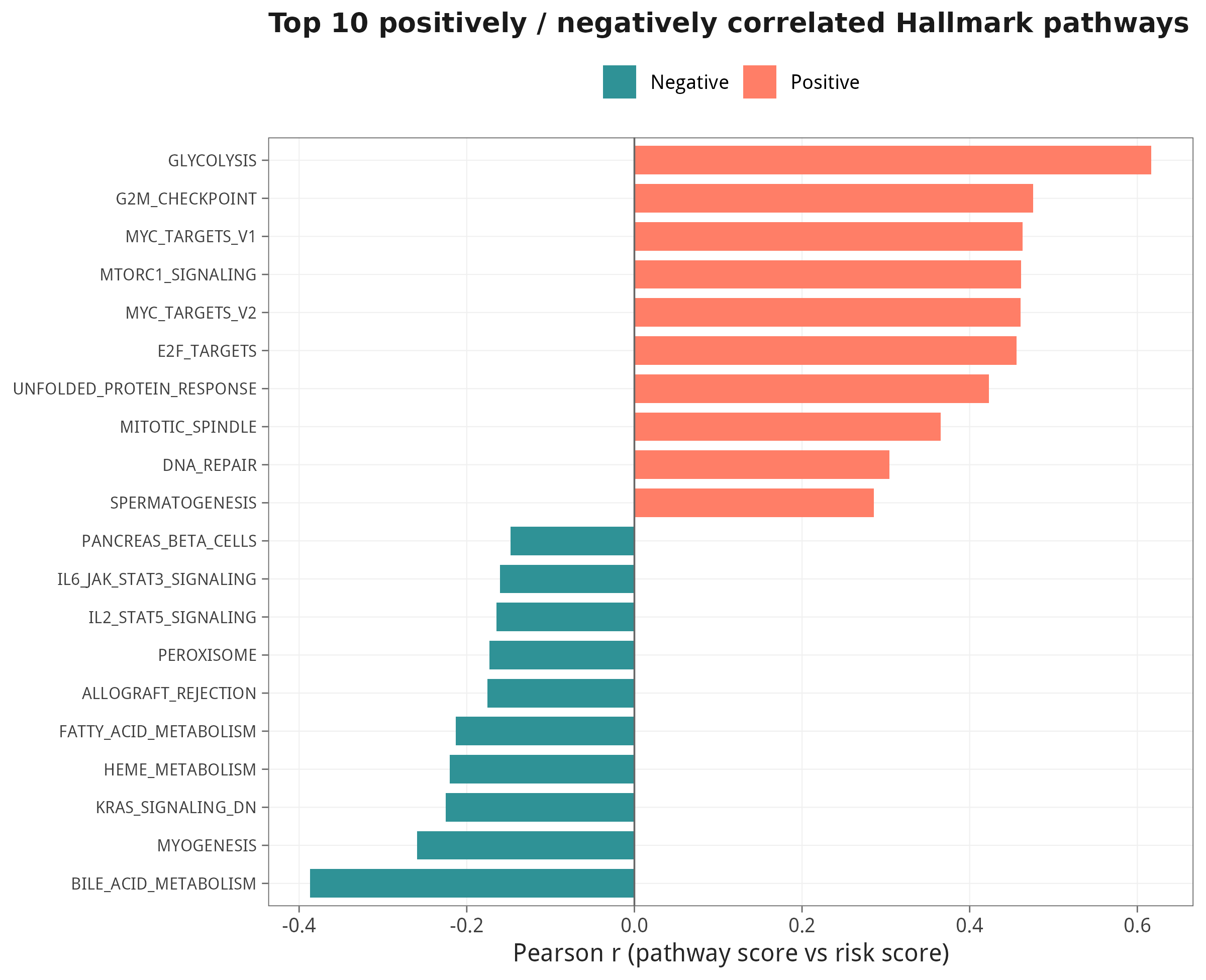
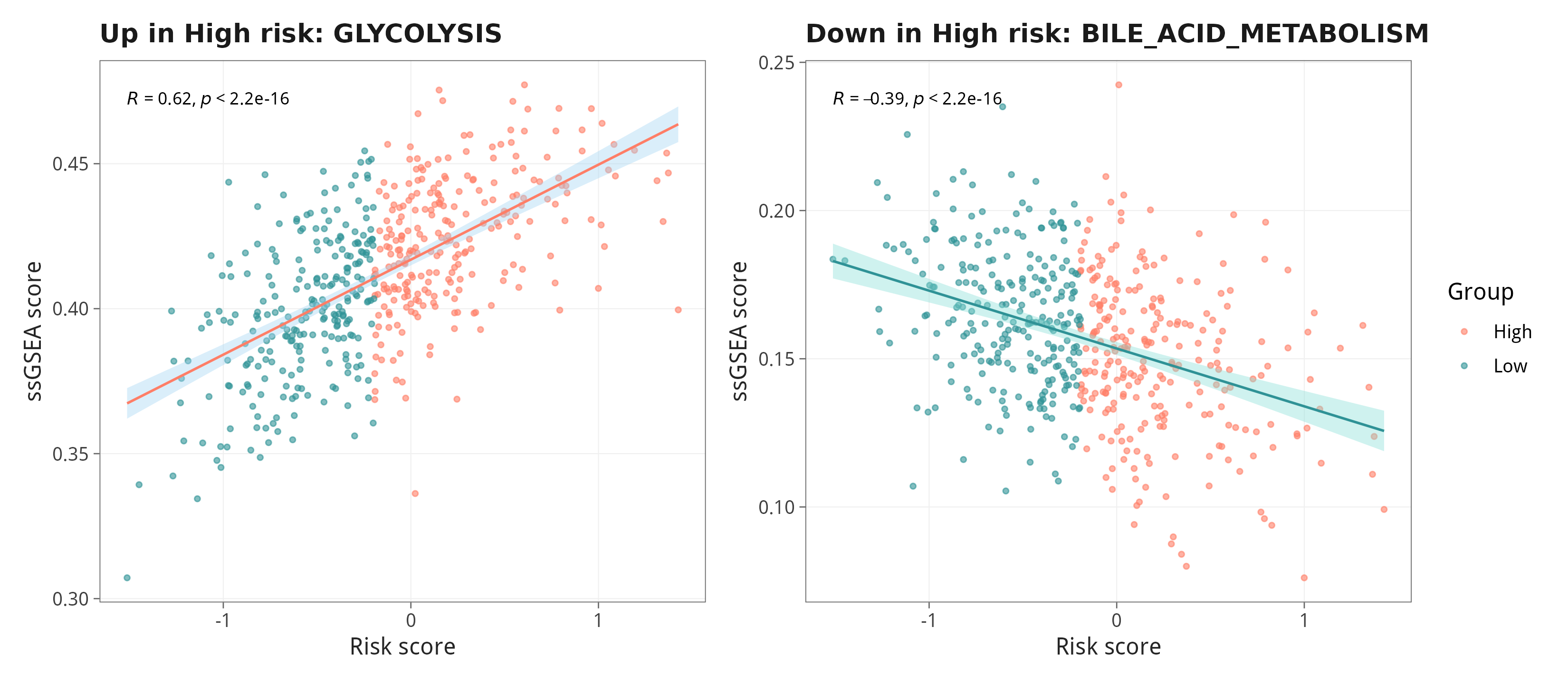
8.4 两条代表性通路的散点示例
第九步 · 分子亚型(ConsensusClusterPlus)
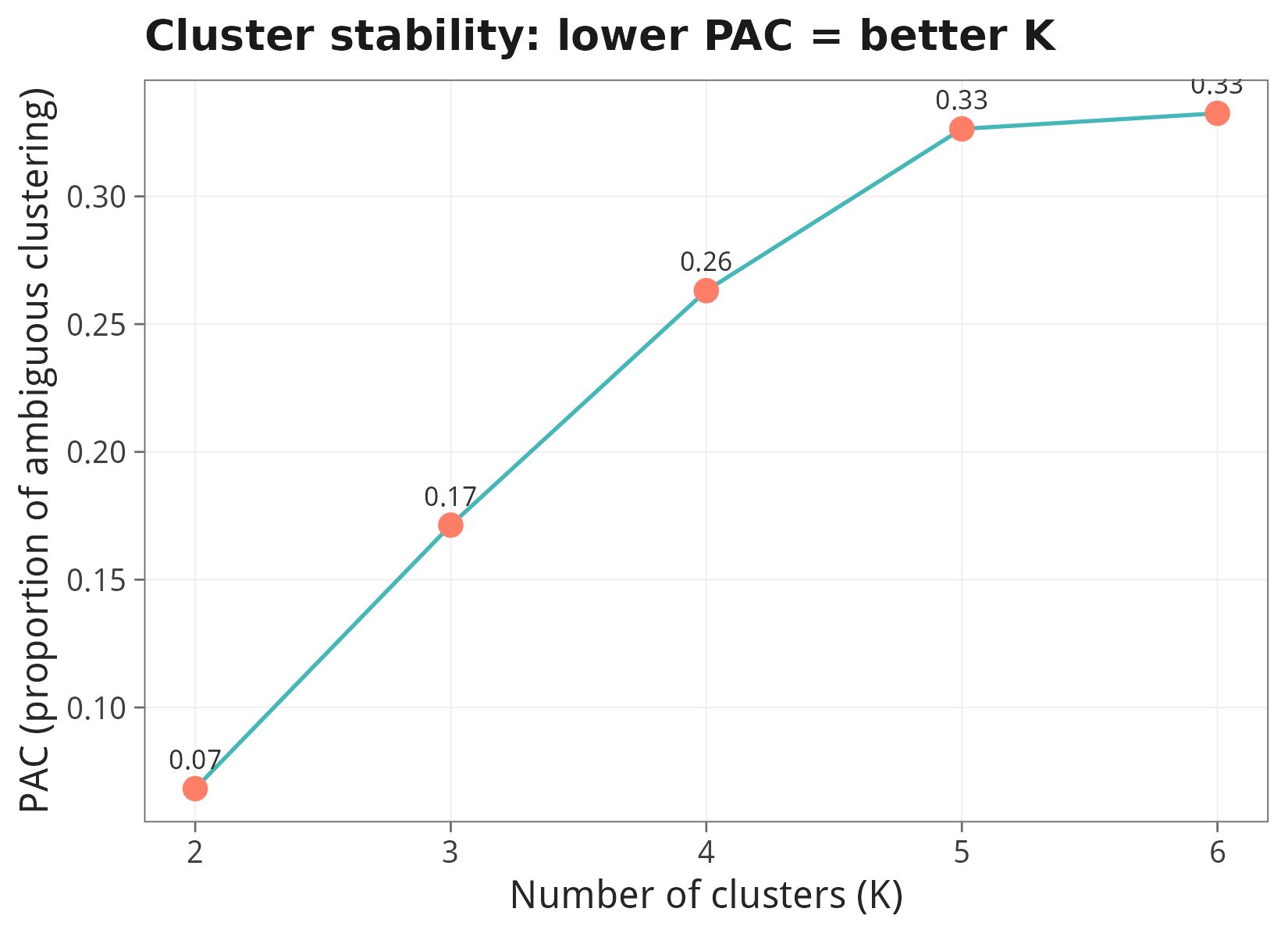
前面的工作都是『把 risk score 作为先验来分组』。本节反其道而行:完全不看生存信息,只用预后基因(uni-Cox p<0.01)的表达做无监督共识聚类,看 LUAD 患者会自然分成几个分子亚型,再回头与 risk group / 生存 / Stage 做关联。
9.1 最佳 K:PAC (proportion of ambiguous clustering)
9.2 亚型 KM 生存曲线
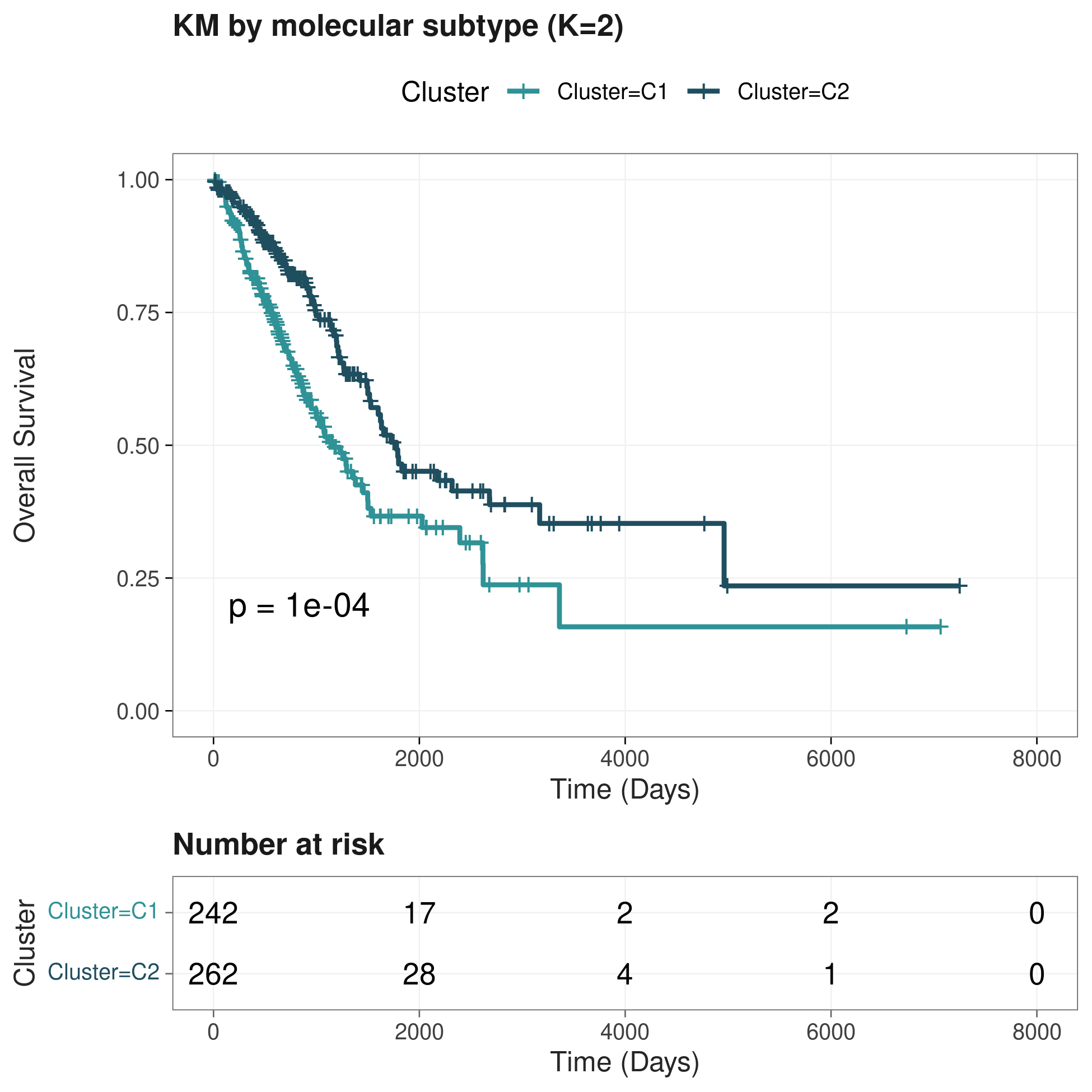
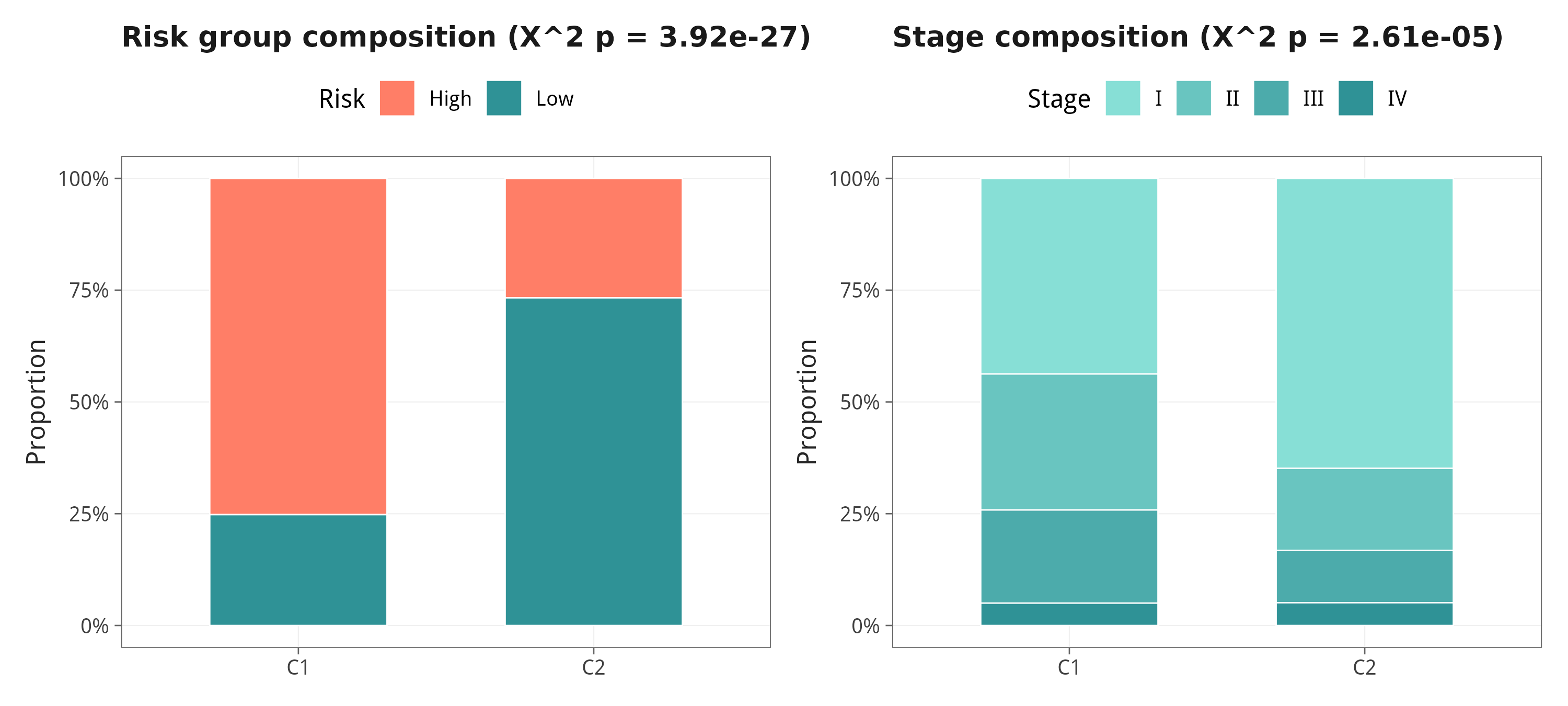
9.3 亚型与 risk group / Stage 的组成关系
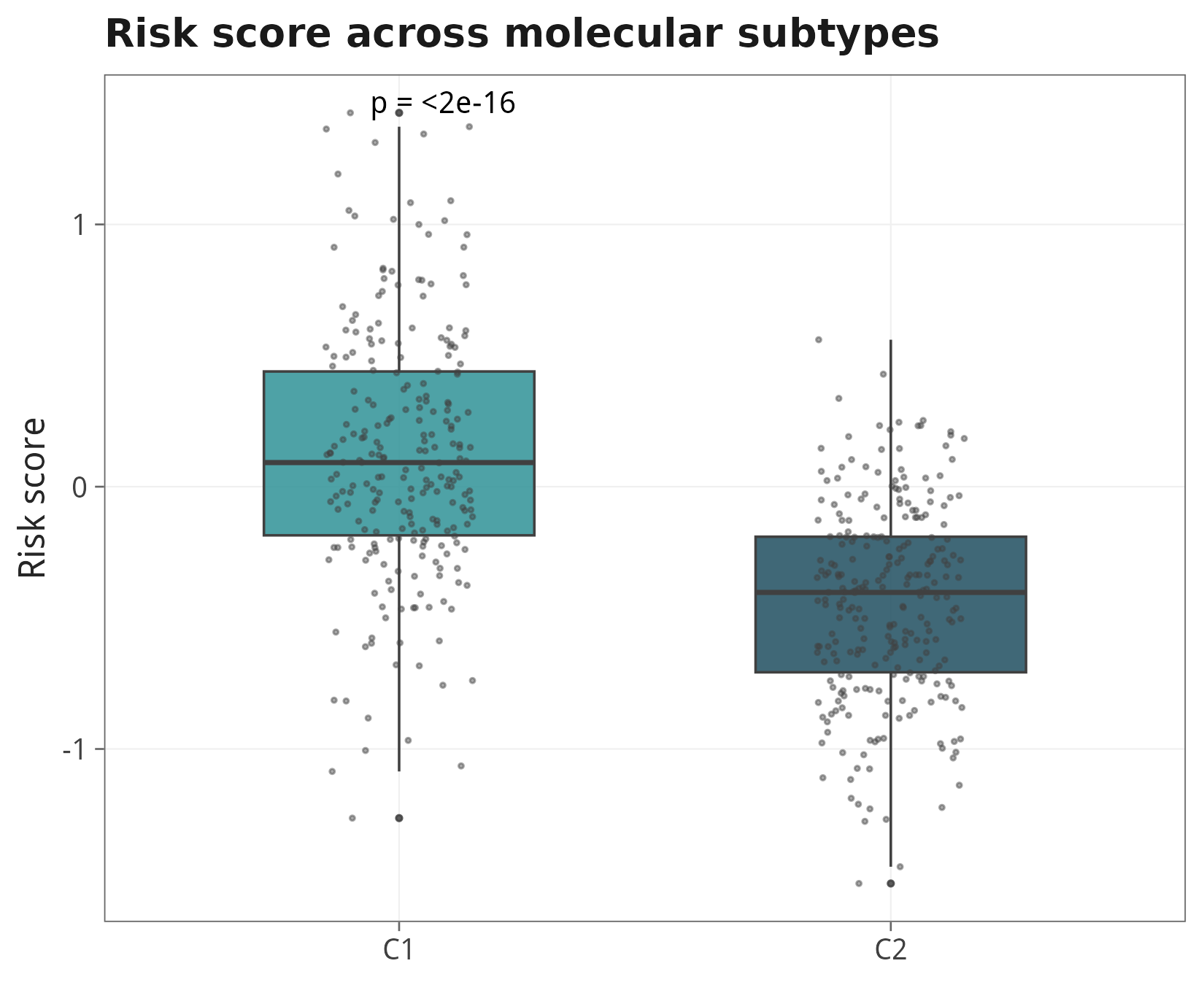
9.4 亚型间的 risk score 差异
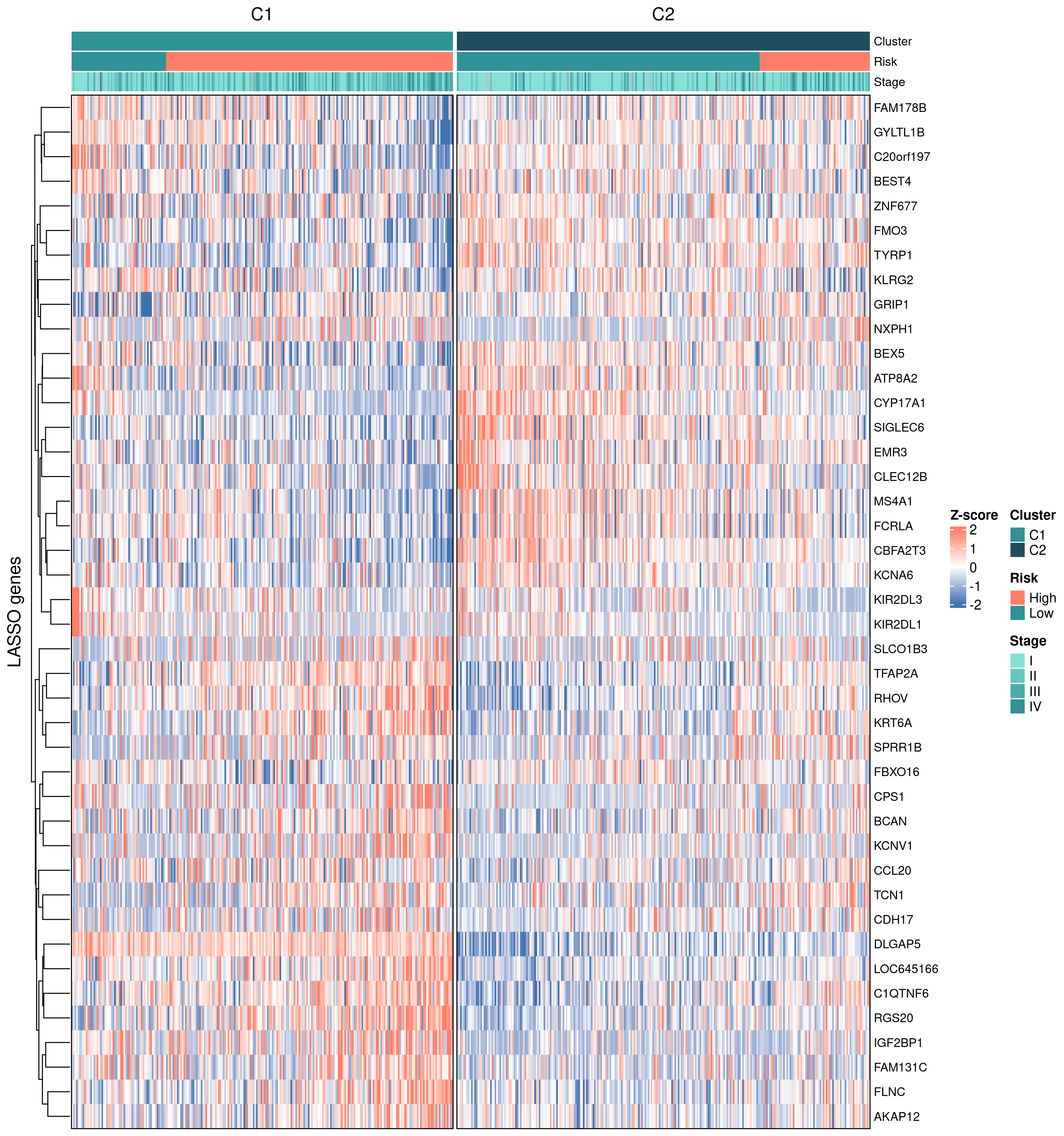
9.5 亚型 × LASSO 基因热图
第十步 · 模型可信度:C-index / DCA / 随机生存森林
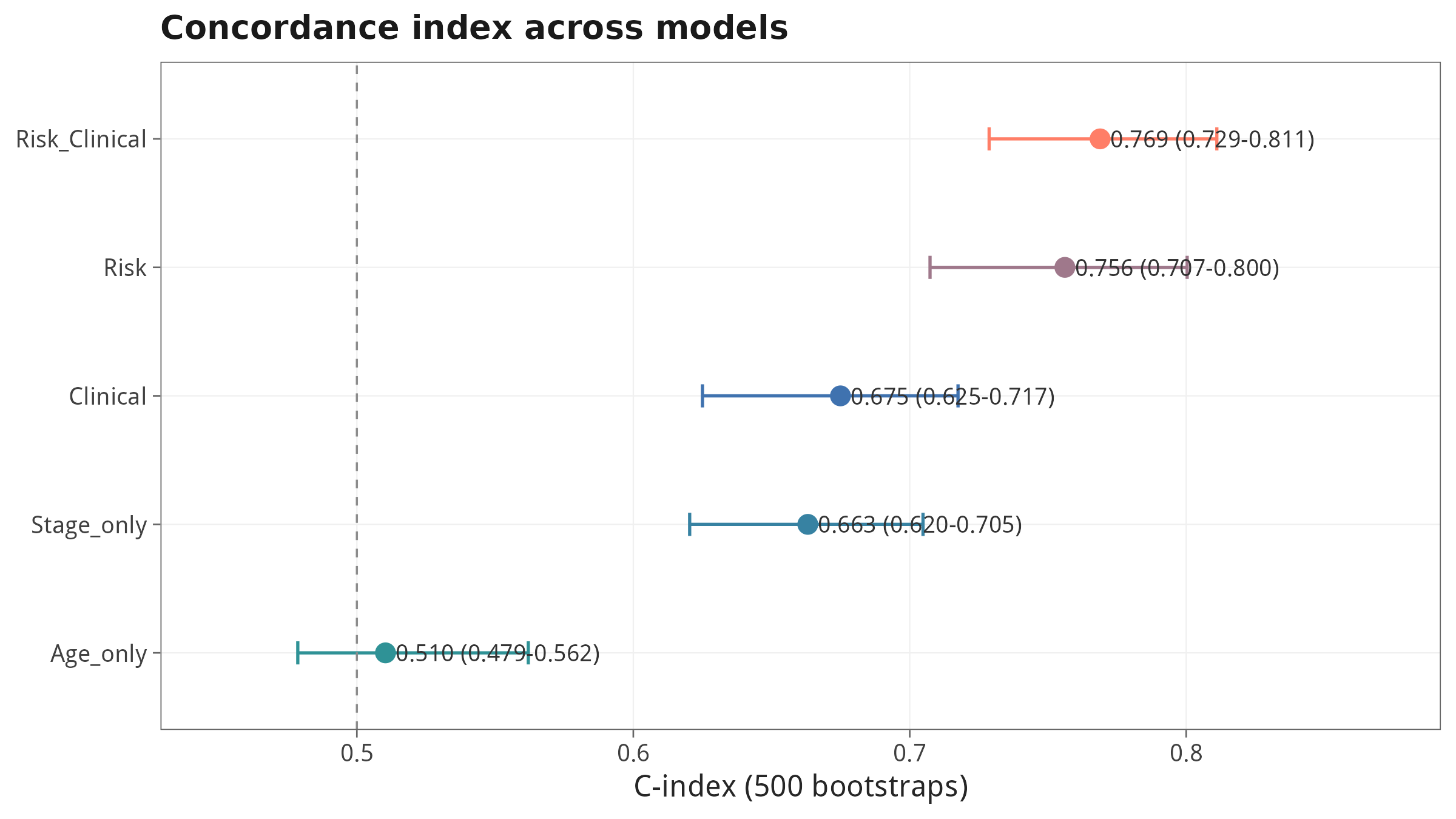
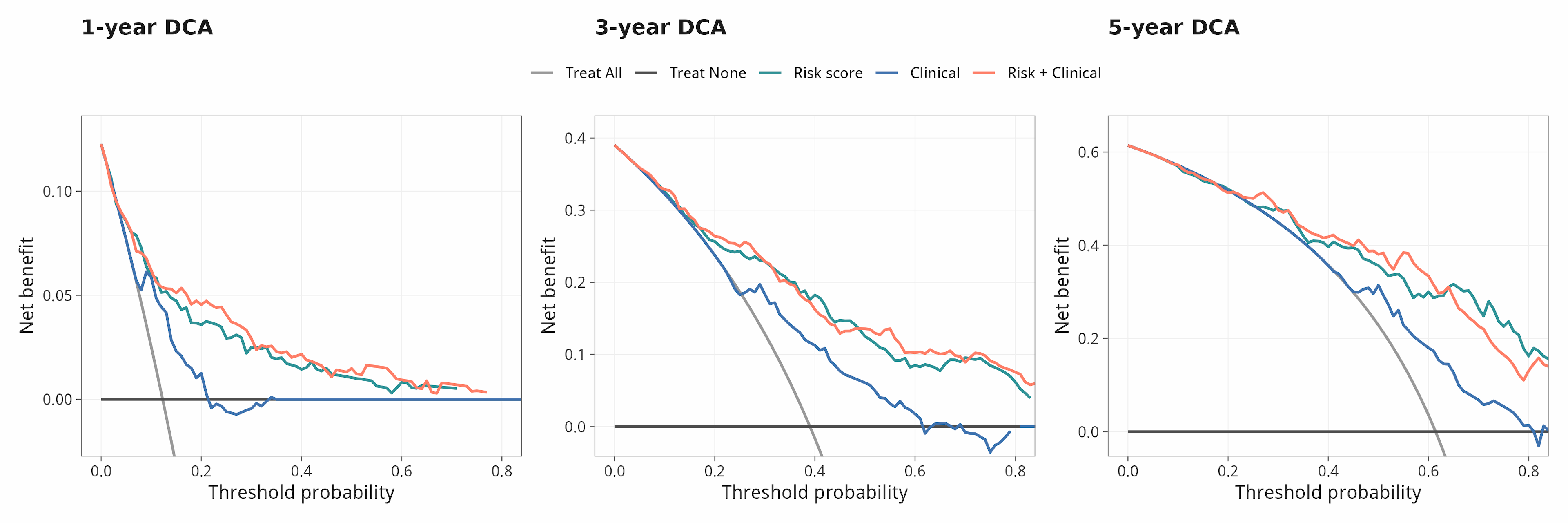
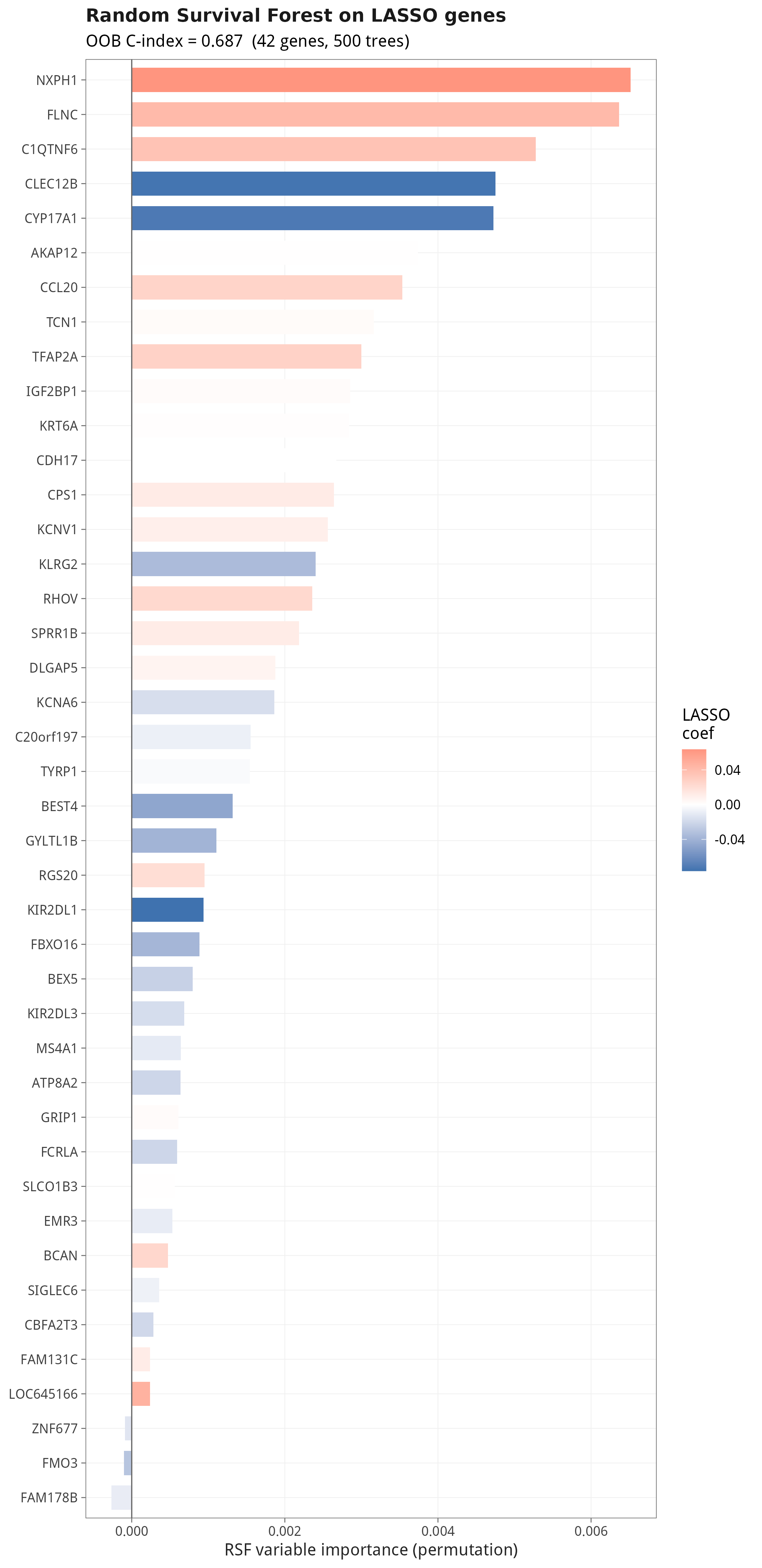
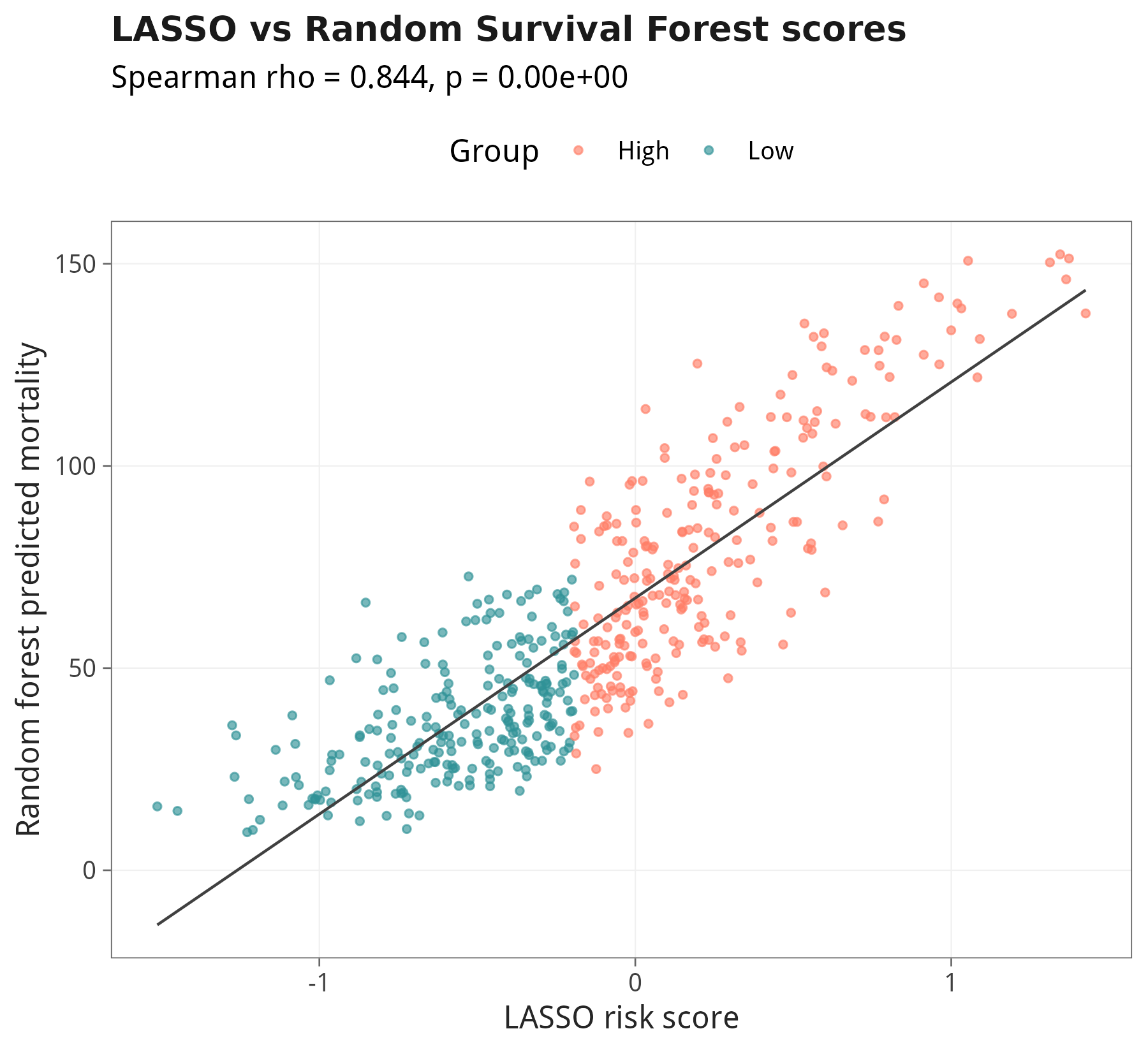
审稿人最常问的三个问题:① 你的模型相对临床变量到底加了多少 predictive power?② 在『临床决策阈值』上有没有 net benefit?③ 用一个完全不同(非线性)的算法回测,13 个基因还是 Top 重要变量吗?本节用 C-index 自助法 + DCA + 随机生存森林(RSF)一次性给出答案。
10.1 C-index 自助法 500 次:5 个模型对比
10.2 决策曲线分析 (DCA, 1 / 3 / 5 年)
10.3 随机生存森林:非线性反向验证 LASSO
10.4 LASSO score vs RSF score 的一致性
第十一步 · miRNA-mRNA 调控网络
miRNA 通过结合 mRNA 3'UTR 抑制翻译 —— 因此一个『真实』的 miRNA-mRNA 调控对应当具备三个特征:① 二者在肿瘤 vs 正常中方向相反;② 在多个权威数据库中有实验验证的结合证据;③ 在同一队列里表达呈显著负相关。本节我们用 multiMiR 拉取 miRTarBase / TarBase / miRecords 的验证数据,经过三重筛选最终保留 5955 条高置信 miRNA-mRNA 边。
mm <- get_multimir(mirna = top_mir, table = "validated",
legacy.out = FALSE)
# Keep validated + opposite DE direction + tumor r < -0.15 (FDR<0.05)
net <- filter_validated_opposite_negative(mm, mrna_deg, mir_deg, expr)11.1 Hub miRNA:靶基因最多的 Top 15
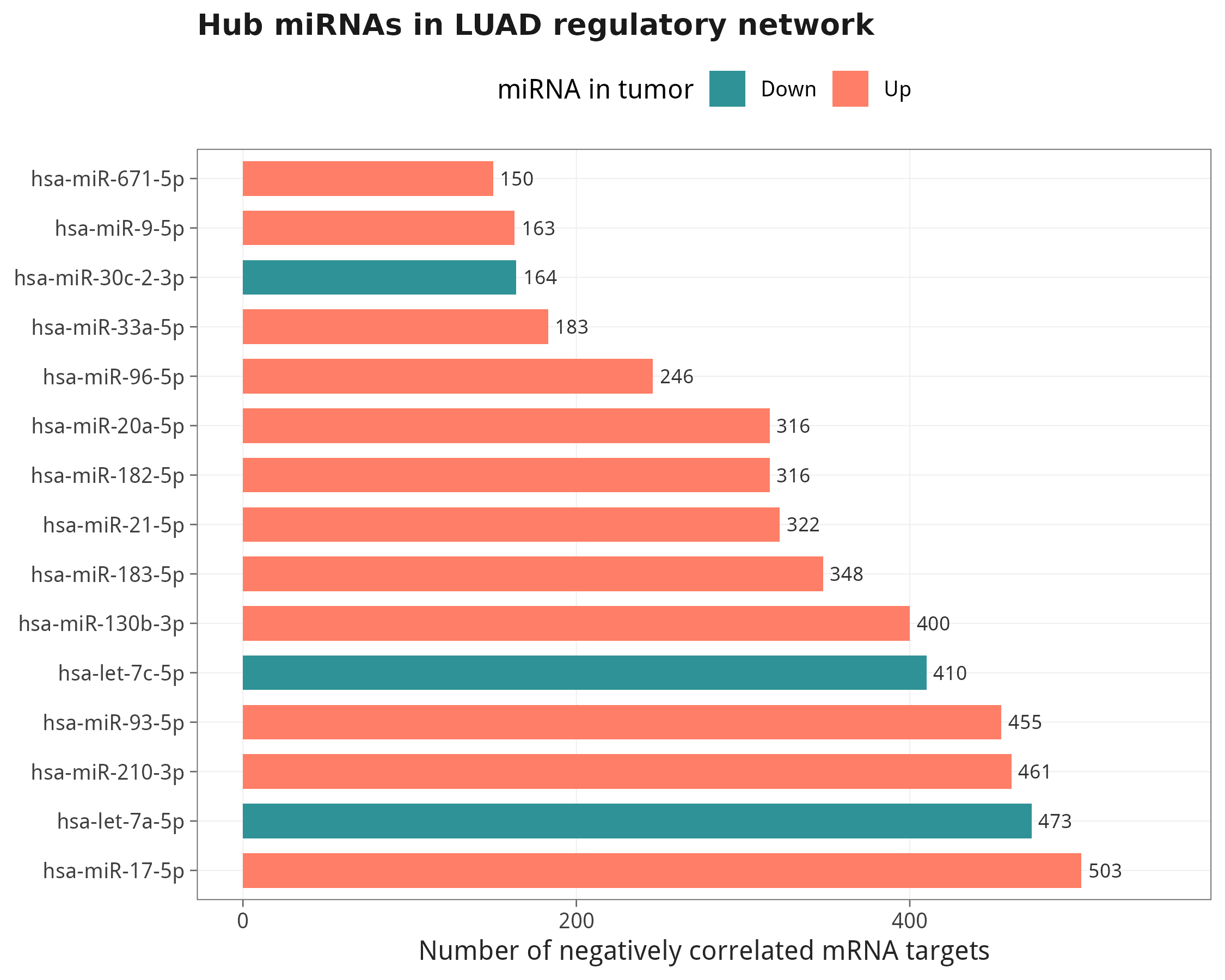
11.2 Top 8 hub miRNA 的可视化网络

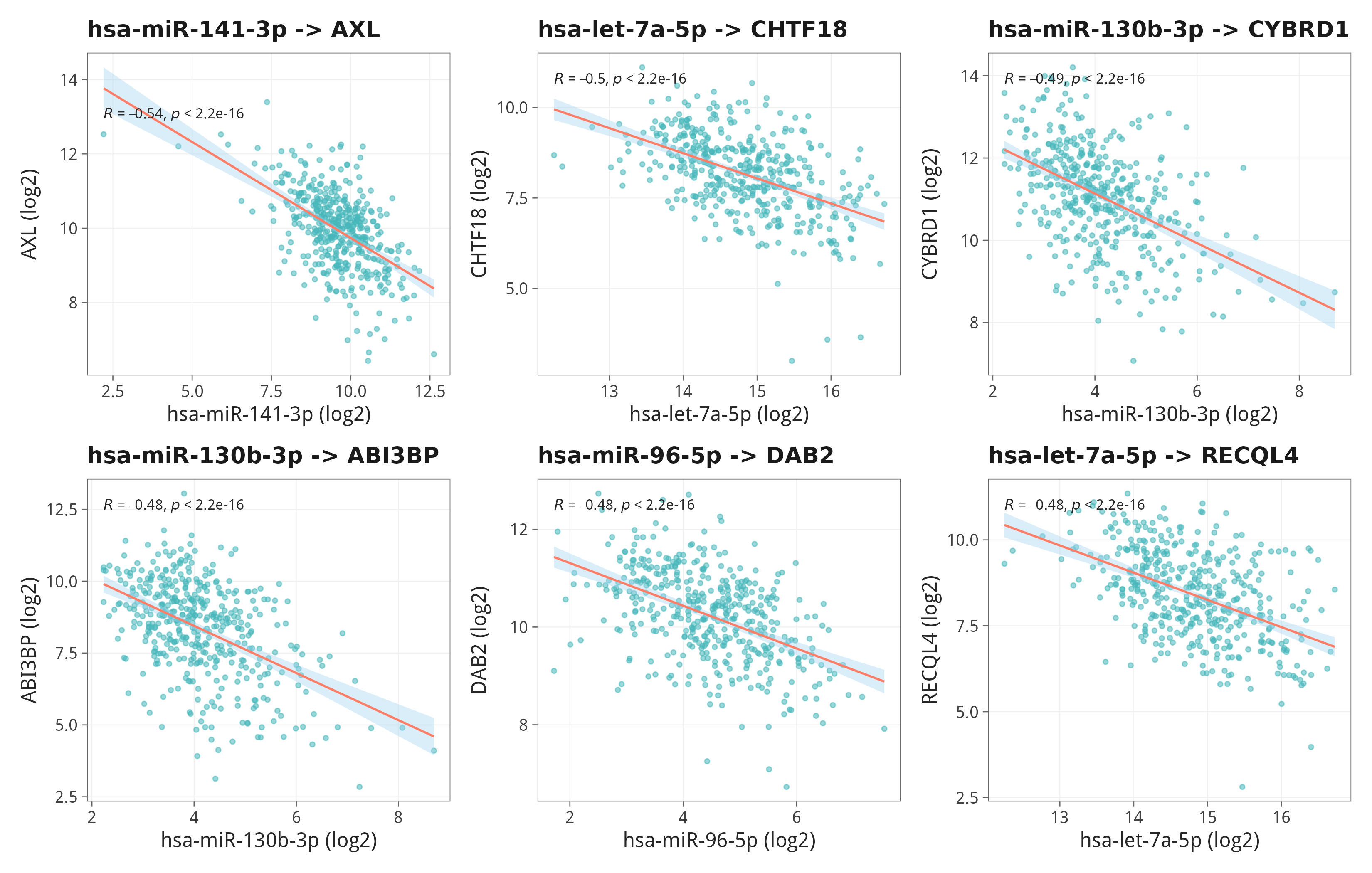
11.3 Top 6 高置信单对的散点验证
第十二步 · 驱动基因突变景观 + 样本流向图
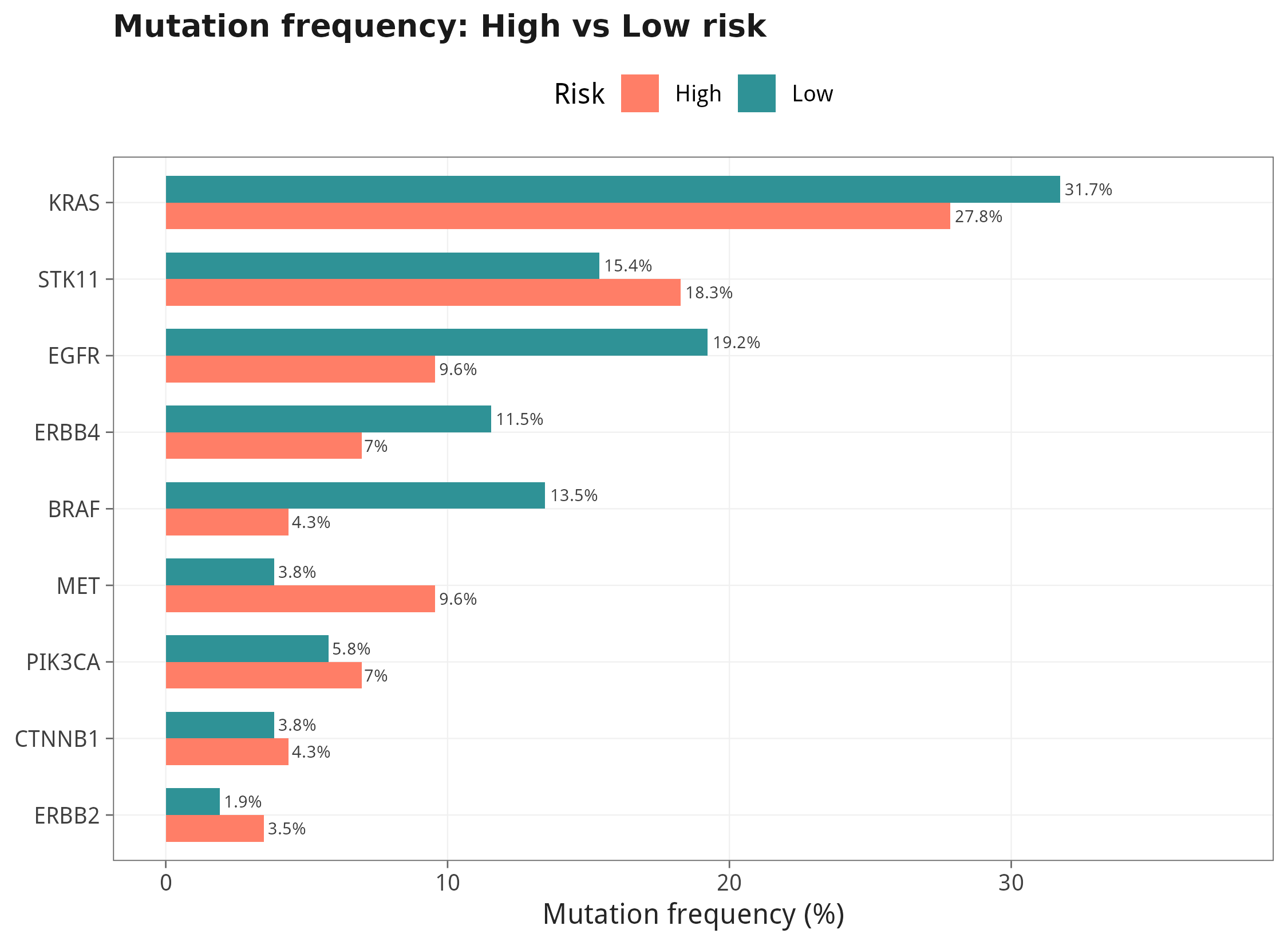
TCGA 临床矩阵自带了 18 个 LUAD 高频驱动基因的氨基酸级注释(KRAS、EGFR、BRAF、STK11、PIK3CA、…),我们把 'none' 编码为 WT、其他编码为 MUT,形成最小可视化的突变景观;并用 Fisher 检验比较高/低风险组的突变频率。
12.1 OncoPrint:突变 + 临床 + LASSO 表达 一张图

12.2 突变频率:高 vs 低风险组
12.3 Sankey / Alluvial:从 Stage 到生死结局的患者流

下篇总结 · 一份范式的迁移指南
把上下两篇拼起来,整套 LUAD 工作流是 12 个 R 脚本 + 73 张图 + 26 个结果表,可以在 504 例 TCGA-LUAD 样本上端到端跑完。故事的主线是:
① 从 4430 个肿瘤 DEG 精炼到一个 13 基因的 LASSO 预后评分;
② 风险评分与 Stage / TNM / 吸烟史显著关联,是多因素 Cox 中的独立预后因子;
③ WGCNA / 免疫浸润 / Hallmark 通路三种不同维度都印证了『高风险 = 增殖与糖酵解上调 + 免疫冷化 + 肺正常功能丢失』;
④ 无监督分子亚型 + 随机生存森林 + DCA 三条独立路径互相确认,整体可信度强;
⑤ miRNA-mRNA 网络 + 驱动突变 OncoPrint 把『分子→通路→预后』的因果链路全部串起来。
整套脚本结构清晰(00_setup / 01-05 基础 + 06-12 进阶),换一个癌种(COAD / STAD / HNSC …)只需要换原始矩阵路径 + 改预处理脚本的几行字段名,其余 11 个脚本几乎零修改即可复用。